
来源:Nature 606,1036-1038(2022)
标题:How to make spatial maps of gene activity — down to the cellular level
作者:Michael Eisenstein
doi:https://doi.org/10.1038/d41586-022-01743-7
计算和实验方法让研究人员准确揭示每个基因在细胞或组织中的表达位置。
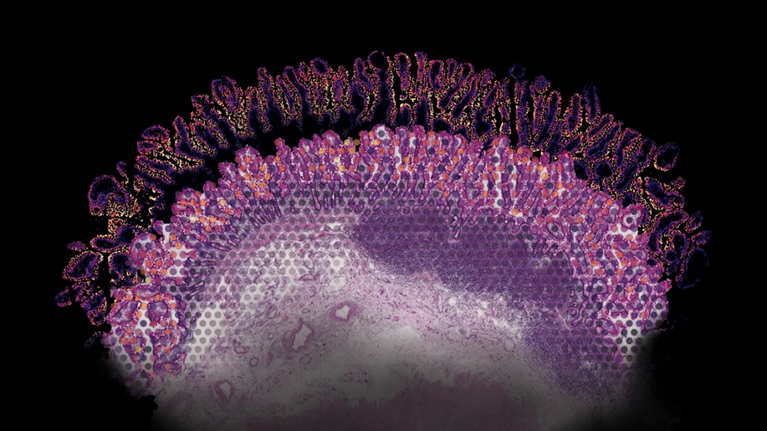
在人类肠道组织的染色切片上,使用基于阵列的空间转录组学方法测量的基因表达数据。来源:Ludvig Bergenstråhle
在显微镜下,哺乳动物组织展现了它们复杂而优雅的结构。但如果你在肿瘤形成后观察相同的组织,你看到的是混乱。纽约大学 Grossman 医学院的计算生物学家 Itai Yanai 试图在这种混乱中寻找秩序。“事物的排列方式有一种特殊的逻辑,而空间转录组学正在帮助我们看到这一点,”他说。
“空间转录组学”是一个笼统的术语,涵盖了十多种用于绘制组织样本中基因表达模式(全基因组)的技术,这些技术是为了补充单细胞 RNA 测序技术而开发的。然而,这些单细胞测序方法有一个缺点——它们可以快速分析大量单个细胞的mRNA 含量(或转录组),但通常需要对原始组织进行物理破坏,这会丢失细胞排列的信息,以至于在后续的分析中出现混乱的结果。以色列雷霍沃特 Weizmann 科学研究所的免疫学家 Ido Amit 说,这样的实验有时会让他的团队质疑实验结果。“这真的是原始位置的状态,或者我们只是在看一些不是主要甚至根本不真实的东西?”
相比之下,空间转录组学使研究人员能够研究完整样本中的基因表达,开辟癌症研究的前沿技术,并揭示现有技术无法表征的生物学现象。由此产生的空间信息“图谱”可以告诉科学家每个组织由哪些细胞构成,它们是如何组织的以及它们如何交流。但编译这些图谱并不容易,因为空间转录组学方法通常存在两个相互竞争的目标:更广泛的转录组覆盖范围和更严格的空间分辨率。实验和计算方法的发展现在正在帮助研究人员平衡这些目标,并在此过程中提高细胞分辨率。
扩大荧光原位杂交的规模
空间转录组学的起源可以追溯到1960年代原位杂交的发展。该技术使用标记的核酸片段作为探针来检测细胞或组织中互补 DNA 或 RNA 序列的存在和位置。最初,研究人员使用放射性标记,但后来转向可以在显微镜下成像的荧光标记。
到1998年,由于显微镜和图像处理技术的进步,研究人员可以识别细胞中的单个 RNA 分子。使用这种单分子荧光原位杂交 (smFISH) 方法,不同颜色的探针同时标记来自多个基因的单个 mRNA。但早期版本的单分子荧光原位杂交一次只能监测三个或四个基因——远低于人类转录组中表达的数万个基因。哈佛大学和麻省理工学院Broad 研究所(马萨诸塞州,剑桥市)的细胞生物学家Fei Chen 说:“显微镜的一个缺陷是你不能一次看到那么多颜色或分子,即使你得到了非常丰富的空间信息。”
此后,对该技术的巧妙改造已经克服了这些限制。例如,生物物理学家庄小威和她的同事在2015年在哈佛大学报告的多重抗错荧光原位杂交 (MERFISH) 可以仅使用几个荧光标签检测和区分来自不同基因的数千个 mRNA。每个mRNA都被分配了一个由1和0组成的唯一二进制条形码,然后用多个包含读出序列的互补“编码探针”进行标记。然后,样品经过连续轮次的杂交和成像,使用各种荧光标记的“读出探针”来破译这个条形码。
当读出探针与编码探针的读出序列结合并发出荧光信号时,它被读取为“1”;如果没有荧光,则读作“0”。多轮成像产生一个二进制条形码,可以识别检测到的 RNA。该技术的“抗错误”部分是指条形码的设计:它们彼此之间有很大的不同,因此几乎不可能误解正在被检测的 mRNA 序列。
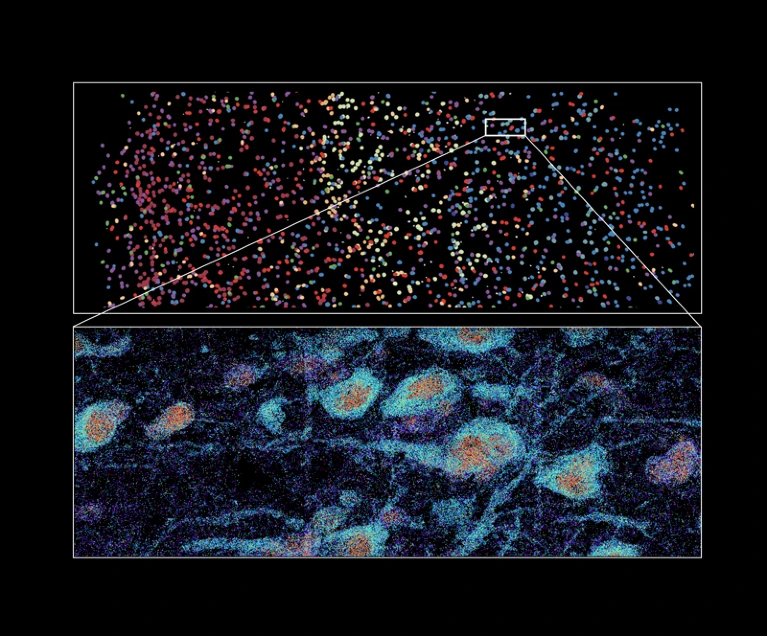
人脑部分的 MERFISH 成像,显示用各种颜色标记的细胞类型(上)和来自单个细胞中不同基因的标记的 RNA 分子(下)。图片来源:庄小威实验室、哈佛大学和HHMI。
尽管该方法最初被描述为单细胞分析的工具,但庄小威的团队也将其应用于组织,包括人脑。“通过分析4,000个基因的表达,我们能够以前所未有的分子和空间分辨率生成分子定义和空间分辨的人类皮层细胞图谱,”她说。这项已发表在《科学》杂志上的分析确定了100多种不同细胞亚型的种类和位置,并揭示了人类与小鼠大脑皮质结构的细胞组成和组织的显著差异。在早期的工作中,庄小威的团队还使用该技术绘制了小鼠大脑的一些部分,包括运动皮层和下丘脑。
其它条形码和成像方法也有类似的优势。例如,2018年加州斯坦福大学的一个团队提出了空间分辨转录扩增子读出映射 (STARmap) ,使用一种原位测序形式来检测完整组织样本中的 mRNA。利用一组基因特异性条形码,每个条形码由 5 个核苷酸组成,斯坦福团队以单细胞分辨率绘制并量化了小鼠脑组织中的 1000 多个基因转录本。
但基于成像的方法也有缺点。例如,由于这些方法囊括更多目标RNA,它们需要很多实验操作。MERFISH一次可以检测超过10,000 个基因,但这种规模的实验通常需要一个额外的步骤——“组织扩张步骤”来扩大每个样本的体积,以便显微镜可以成功地解析不同的分子。另一种方法 seqFISH+ 通过使用更复杂的颜色编码策略克服了这一限制。但是对于相同数量的基因,seqFISH+需要更多轮的标记和成像——80轮,而MERFISH需要23轮。这两种方法都需要一天以上的不间断的时间来用显微镜收集全转录组规模的数据。
一系列替代方案
基于杂交的技术最基本的限制应该是研究人员必须提前决定他们希望标记哪些基因。“一旦你开始选择目标基因,你就会丢失信息,”Amit 说。基于阵列的方法提供了更广泛的转录组视图,但需要付出一定的代价——它们的灵敏度较低,空间分辨率也较低。
JoakimLundeberg是斯德哥尔摩KTH皇家理工学院的分子遗传学家,他是空间转录组学行业先驱之一。他和他的同事在载玻片上排列点状的核苷酸探针阵列,旨在捕获 mRNA单链。这些通过与终止每个 mRNA 转录的腺嘌呤核苷酸长尾结合来发挥作用。在将薄薄的组织切片放在载玻片上后,研究人员用试剂处理组织,使其具有穿透性,使暴露出RNA并与阵列结合。然后将捕获的 RNA 转化为 DNA 并测序。因为每个核苷酸探针都包含一个独特的条形码,表明它在载玻片上的位置,最终数据不仅揭示了 mRNA 的序列,还揭示了它在组织中的位置。然后可以将生成的数据可视化为覆盖在显微图像上的像素化图,其中每个像素显示在每个位置表达了哪些基因。
Lundeberg 的团队已经使用这种技术对大脑和肿瘤组织样本的完整转录组进行了采样,尽管空间分辨率有限。在最初的方法中,像素描述的点直径大约为100微米——比典型细胞大10倍。从那时起,该技术已被加利福尼亚州Pleasanton的10xGenomics公司商业化,即Visium空间基因表达平台,像素大小为55微米。Yanai 的团队使用该平台绘制了胰腺和皮肤肿瘤的结构图。他说,即使没有单细胞分辨率,他们也获得了关于肿瘤结构和癌细胞、健康宿主组织和免疫细胞群之间重要生物学相互作用的重要信息。
在过去的几年中,人们为提高基于阵列的方法的分辨率做出了一系列努力。例如,Chen和他在Broad研究所的合作者EvanMacosko开发了一种名为Slide-seq的方法,其分辨率为10微米——大约是单个细胞的大小,Chen 说。10x Genomics 宣布其将于今年晚些时候发布的下一代 Visium HD 平台也将提供单细胞分辨率,尽管迄今为止尚未公布任何数据。
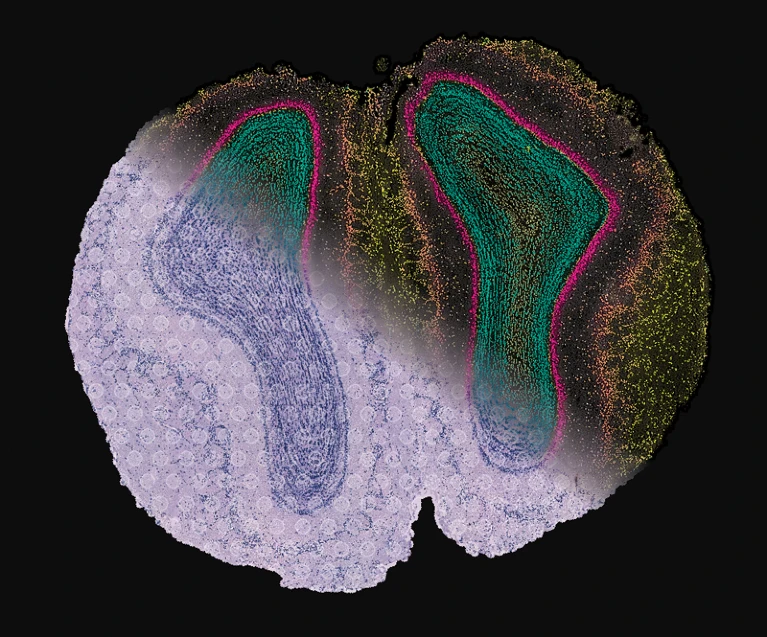
组合图像显示了小鼠嗅球中细胞的组织结构、RNA 数据和超分辨率基因表达图。来源:Ludvig Bergenstråhle
今年5 月,位于中国深圳的生命科学公司华大基因(深圳)的研究人员提出了一种基于阵列的方法,可以突破单细胞分辨率。它被称为 Stereo-seq,它使用带条形码的 DNA 纳米球的图案阵列,这些纳米球的直径大约为 200 纳米,相距数百纳米。“我们实际上有大约 400 个数据点来生成一个细胞,”BGI 集团执行董事、该方法的开发者之一徐迅说。它可以应用于大型样本,包括整个猕猴的大脑被切成三乘五厘米的切片,正如今年的预印本中所报道的那样。华大基因(深圳)的陈说,仅测序就花了将近两个月的时间,他也是 Stereo-seq 团队的一员。
但随着分辨率的提高,技术挑战也随之而来。一种是扩散:当 mRNA 从组织中逃逸出来时,它们会在遇到捕获探针之前横向扩散,从而改变数据。Lundeberg 说,通过优化组织的穿透性,研究人员可以将这种扩散限制在几微米,这对于细胞分辨率来说已经绰绰有余了。“如果你真的想看到亚细胞分辨率,你应该选择基于成像的平台,”他建议道。
另一个挑战是物理学方面的挑战:随着像素大小的减小,可用于捕获 mRNA 的探针数量也在减少。Lundeberg 说他放弃了他团队平台的高分辨率版本,因为它缺乏捕捉生物学相关 mRNA 信号的灵敏度。华大基因团队报告说,Stereo-seq通常可以检测每个细胞300-500个基因,这提供了一个有用但数量有限的基因表达活动视图。尽管如此,该团队还是使用该方法构建了 3D 图谱,该图谱绘制了小鼠、苍蝇和斑马鱼的胚胎发育过程中基因表达的空间变化。
解读每一行
解读空间数据需要专门的计算工具。例如,研究人员可能需要使用仅对转录组的一个子集进行采样来推断细胞类型。许多研究人员通过平行分析从同一组织收集的单细胞 RNA 测序数据来实现这一目标。“然后,您可以将您在空间数据中看到的内容与您在单细胞数据中看到的内容进行匹配,”Fei Chen 说。这种对比方法使研究人员将从 RNA 测序数据集推断的细胞类型定位到空间转录组图上。
一些算法甚至可以计算出由 Visium 等平台产生的相对较大像素的细胞结构,该平台可以解析多个细胞。Fei Chen 和哈佛大学的计算生物学家 Rafael Irizarry 为这种分离过程开发了一种开源算法,称为高效细胞类型分解 (RCTD),也称为点反卷积。RCTD 广泛适用于大多数基于阵列的方法,Fei Chen 说。它不仅可以识别哪些细胞存在于给定像素中,还可以揭示缺失的细胞基因表达活动的细节。RCTD 可以应用于基于成像的方法,例如用于MERFISH图像分割,Fei Chen 补充道——从来自单细胞 RNA 测序的基因表达数据中识别细胞边界。
费城宾夕法尼亚大学的遗传学家和统计学家Mingyao Li说,成像数据也可以成为细胞去卷积的重要结果,大多数基于阵列的空间转录组学技术可以同时捕获此类数据。“你可以放大,你可以查看组织特异性特征,有多少细胞,细胞密度是多少,以及单个细胞的形态特征是什么,”她说。但是将这些元素联系在一起是一项具有挑战性和数据密集型的任务,通常需要复杂的计算方法。
例如,Lundeberg 及其同事发表了一项研究,在该研究中,他们使用来自 Visium 仪器的转录组学和组织学数据训练了一种深度学习算法,以推断出单个采样点更多的细节。“我们可以非常准确地预测点与点之间的基因表达,”他指的是每种基于阵列的方法所固有的物理间隙。“我们实际上可以从中推断出单细胞分辨率。”
然而,识别细胞类型仅仅是开始。不同的细胞类型可能具有明显不同的表型,具体取决于它们在组织中的位置,这些差异基因表达模式可以使空间细胞图谱更加强大。机器学习算法对于梳理这种多样性也很有用。例如,Amit 及其同事开发了一种称为 DestVI 的技术,该技术既可以解析每个阵列点中的细胞种类,又可以捕获各种细胞类型中的独特生物状态。使用这个技术,该团队确定了癌组织中的免疫细胞表型。“人们可以对组织的生理学或病理学有更高层次的理解,”Amit说。
把这一切结合在一起
对于一个产生如此多数据的领域来说,也许会令人惊讶,空间转录组学研究人员现在需要更多的数据。人类细胞图谱(HumanCellAtlas)等倡议特别重要,该图谱发布了从33个器官的数百万个细胞中收集的转录组数据(www.humancellatlas.org )。例如,这种高质量的标准化数据可用于训练分析算法。
空间转录组学尚未达到基因组学或单细胞转录组学等更成熟领域所拥有的协作和数据共享水平,这可能是一个缺陷。Fei Chen 说,在许多情况下,实验室只会分享出版商和资助者要求的最低限度——来自实验的原始、未经处理的数据——这意味着重复这些工作可能需要几个月的时间。但目前已经出现了有希望的进展。例如,在其 Stereo-seq 工作发表后,华大集团启动了时空组学联盟,该联盟已经吸引了来自世界各地的 80 多名研究人员。其目标是使用各种空间方法来解决与人类生理学、发病机制和进化生物学相关领域的难题。
与此同时,研究人员正在寻求进一步增强该技术。例如,Lundeberg 的团队正在使用空间转录组学来推断前列腺肿瘤发展过程中发生的基因组变化——通常只能从分离细胞的基因组测序中获得这些信息。他说:“在单个组织切片中,你会看到这些极早期的事件,这是之前没有人发现的,”他补充说,许多这些变化发生在原本看起来是良性的细胞中。
至于Yanai,他对相邻细胞如何相互交流和相互影响更有兴趣。这种信息交互是正常器官形成和发育的重要组成部分,有助于揭示肿瘤组织的组成原理。“癌细胞正在操纵非癌细胞,”Yanai 说。空间转录组学可以在这种操作发生时捕捉到它。“这就像拼图中缺失的一块,”他说。